Antiphospholipid Syndrome: The Clotting Disorder No One Sees Coming
Antiphospholipid syndrome is an autoimmune thrombophilia: antibodies directed at phospholipid-binding proteins trigger clotting in arteries and veins throughout the body. It is the most common acquired cause of recurrent miscarriage and unexplained stroke in young adults, yet most patients have no warning before the first thrombotic event.
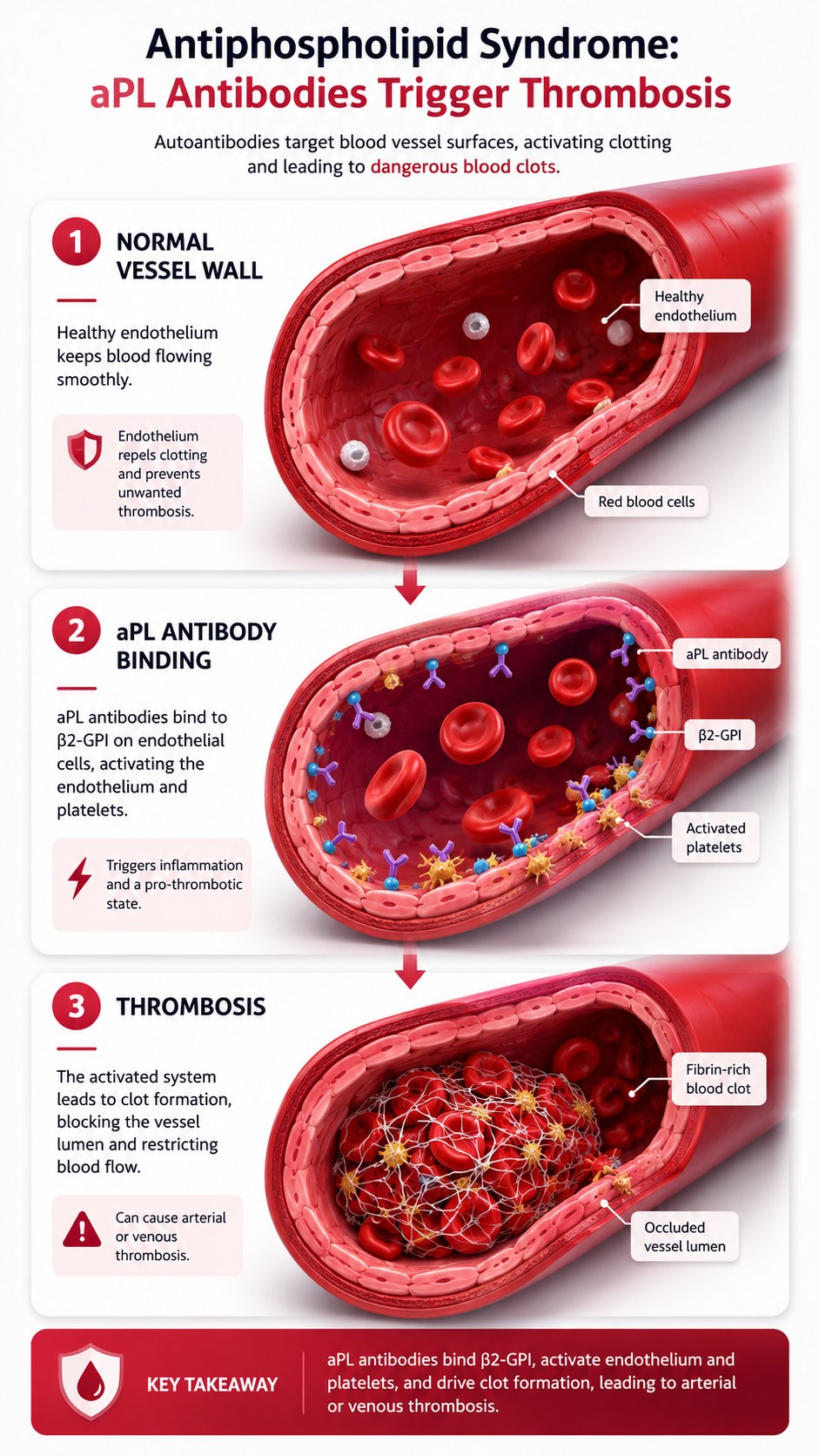
What is happening in the body
Antiphospholipid syndrome (APS) is caused by autoantibodies — antiphospholipid antibodies (aPL) — that target phospholipid-binding plasma proteins, most importantly beta-2 glycoprotein I (β2-GPI) and prothrombin. β2-GPI is a natural anticoagulant protein that binds to phospholipids exposed on cell membranes under conditions of cellular stress. When aPL antibodies bind β2-GPI on endothelial cells, monocytes, and platelets, they trigger an activation cascade: endothelial cells upregulate tissue factor and adhesion molecules; monocytes produce procoagulant microparticles; platelets aggregate. The net effect is a shift from the normal anticoagulant surface of blood vessels to a prothrombotic one.
The clinical consequence is arterial and venous thrombosis — blood clots forming in vessels where none should form. Deep vein thrombosis and pulmonary embolism are the most common venous manifestations. Stroke and transient ischaemic attack are the most dangerous arterial manifestations, particularly in young patients who have no traditional cardiovascular risk factors. In pregnancy, aPL antibodies interfere with placental implantation and perfusion, causing recurrent miscarriage, intrauterine growth restriction, and pre-eclampsia.
The symptoms this produces
- Deep vein thrombosis — swollen, painful, red leg
- Pulmonary embolism — sudden breathlessness, chest pain, collapse
- Stroke or TIA in young adults without obvious cardiovascular risk
- Recurrent pregnancy loss (particularly second-trimester miscarriage)
- Livedo reticularis — mottled, net-like skin discolouration
- Thrombocytopenia (low platelets, paradoxically)
- Catastrophic APS: simultaneous thrombosis in multiple organs — a rare, life-threatening variant
How this fits the autoimmune pattern
Antiphospholipid antibodies are frequently triggered by infection — particularly by Gram-negative bacteria expressing phospholipid-mimicking antigens that prime cross-reactive B-cells, and by EBV and CMV that expose β2-GPI during cellular lysis. The gut is the largest reservoir of Gram-negative organisms, and increased intestinal permeability provides the systemic exposure to bacterial lipopolysaccharide and cell membrane phospholipids that generates aPL antibodies. APS frequently co-occurs with SLE — a relationship that underscores the shared upstream immune activation environment — and shares with lupus the anti-phospholipid/nuclear antigen antibody signature.
What the clinical data shows
APS management typically requires long-term anticoagulation with warfarin or direct oral anticoagulants (DOACs) — drugs that manage the thrombotic risk but do not address the underlying aPL antibody production. Dietary intervention targeting the gut permeability and dysbiosis that drives aPL production is an emerging complement to pharmacological management. Several APS patients in clinical practice have shown declining aPL titres on anti-inflammatory dietary protocols, suggesting that the immunological driver is accessible to dietary modification, even when pharmacological management of thrombotic risk remains necessary.
A life with this condition — Stories
Kezia, 34. She had her third miscarriage, all in the second trimester. Each had been explained individually — chromosomal abnormality, cervical incompetence, suboptimal luteal support. After the third loss, her obstetrician ordered a thrombophilia screen. All three antiphospholipid markers came back positive, confirmed on repeat testing twelve weeks later. The syndrome had probably also caused the brief left arm numbness she had had at 29 and attributed to a trapped nerve. Her immune system had been producing antibodies that turned her blood vessels into clot-forming surfaces. She had been one thrombotic event away from a stroke for years and had not known it.
Oliver, 29. He woke up unable to move his right arm and with slurred speech. In the emergency department, the stroke team worked quickly — clot-busting medication within the window, MRI confirming a left middle cerebral artery territory infarct. He was 29, did not smoke, had normal cholesterol, normal blood pressure, no family history of early stroke. The haematology workup ordered during his admission found lupus anticoagulant and anti-cardiolipin IgG antibodies. He had had a stroke from a condition that had been generating a clot-prone vascular environment for years before anyone thought to look for it.
Sophie, 45. Her APS was discovered after her second DVT in three years, the second of which threw a pulmonary embolism that was found incidentally on a CT scan ordered for chest pain. Her haematologist tested her for thrombophilia and found antiphospholipid antibodies; further investigation found she also had lupus — secondary APS, occurring in the context of another autoimmune disease. She was told she would need anticoagulation for life and was referred to a lupus clinic. She had spent three years being told her DVTs were unprovoked, which had shaped her treatment. They were not unprovoked. They had a cause, and the cause had a treatment.
Miriam, 41. Her APS was catastrophic — the rare variant in which thrombosis occurs simultaneously in multiple organ systems. She presented with stroke, renal thrombosis, and adrenal infarction within 48 hours of what appeared to be a viral gastroenteritis. The trigger for catastrophic APS in her case was the infection activating complement and endothelium simultaneously in a patient whose antiphospholipid antibodies had been present, undetected, for years. She survived after intensive anticoagulation and immunosuppression. Her haematologist told her that catastrophic APS carries a 30-50% mortality rate. She was in the half who lived. She did not know, before that week, that she had the condition at all.
Transcript witness — clinical commentary. On APS and the gut-infection connection: "Antiphospholipid antibodies are frequently triggered by infection — particularly by Gram-negative bacteria expressing phospholipid-mimicking antigens that prime cross-reactive B-cells. The gut is the largest reservoir of Gram-negative organisms, and increased intestinal permeability provides the systemic exposure to bacterial lipopolysaccharide and cell membrane phospholipids that generates aPL antibodies in susceptible individuals. In patients with APS, declining aPL titres on dietary protocols that address gut permeability suggest the antibody production is actively maintained by ongoing microbial antigen exposure — and can be reduced when that exposure is reduced."
Thomas, 33. He had been told he had a hypercoagulable state after his DVT and was put on anticoagulation indefinitely. Three years later, a haematologist who reviewed his records wondered why an antiphospholipid antibody screen had never been done. It came back positive for anti-beta-2-GPI and lupus anticoagulant. He now had a diagnosis rather than a description. The change mattered because APS-associated thrombosis has specific management considerations — in particular, the choice of anticoagulant — that differ from other thrombophilias. He had been appropriately treated. He had not been appropriately diagnosed.
Grace, 29. Her fourth miscarriage led her fertility specialist to finally raise the possibility of antiphospholipid syndrome. The first three losses had been attributed to embryonic chromosomal abnormalities — a plausible explanation for any individual miscarriage but statistically improbable as an explanation for four. Her aPL panel was positive. She was started on low-dose aspirin and low-molecular-weight heparin injections from her next positive pregnancy test. Her fifth pregnancy delivered a healthy baby at 38 weeks. The treatment that had been available since her second miscarriage was prescribed only after her fourth. The delay had a cost she carried for a long time.
Harriet, 55. She had managed APS for fifteen years on warfarin when the target INR range of 2.5-3.5 her haematologist had set — higher than the standard range, reflecting the arterial thrombosis component of her disease — began to be questioned by a new haematologist who favoured a lower range. A trial of a lower target produced a TIA at three months. She returned to the higher range. What she had learned from the TIA was that her disease had opinions about her INR that were more authoritative than the literature's uncertainty about the optimal target. She had managed herself with the higher range for fifteen years without incident. That was the data. She trusted it more than the new haematologist's preference.